초파리는 단순한 생물이지만 유전자 연구에서 놀랍도록 강력한 모델 생물로 알려져 있습니다. 그렇다면 이 작은 곤충의 유전자는 과연 인간과 얼마나 닮았을까요? 놀랍게도 초파리와 인간의 유전적 유사성은 매우 높습니다. 초파리의 전체 유전자 중 약 60%는 인간과 유사한 서열 또는 기능을 가지며 특히 암 관련 유전자에서는 매우 높은 유사성을 보입니다. 유전적 유사성 뿐만 아니라 해부학적 구조도 비슷한데 신경계, 심장, 식도, 장, 생식선, 지방 등과 같이 인간이 가지고 있는 조직 및 기관과 구조적 및 기능적 유사한 면을 가져 치료제 발굴에 적극적으로 사용되는 모델 생물이기도 합니다 (그림 1A).
대표적인 초파리 기반 암 연구 모델 중 하나는 인간 RET 유전자를 발현한 형질전환체입니다. RET는 세포 증식과 분화에 관여하는 수용체 타이로신 키나아제로 활성화 돌연변이(예: MEN2A, MEN2B, RET/PTC 재배열)는 다양한 종양을 유발하는 것으로 알려져 있습니다. 이를 모사하기 위해 연구자들은 Gal4/UAS 시스템을 이용하여 초파리의 눈 조직에서 돌연변이형 RET (dRet)을 발현시켰으며 그 결과 극심한 안구 과증식과 조직 이상이 나타났습니다 (그림 2). 이러한 형질전환체는 인간 암 유전자의 발암 기전을 생체 수준에서 재현했으며 약물 저해제 (ZD6474)의 효능을 신속하게 평가하였습니다. 실제로 RET 저해제인 ZD6474 처리 시 돌연변이 RET로 인한 안구의 비정상적 발달이 현저히 억제되는 것이 관찰되어 초파리 모델이 암 유전자 기능 연구와 약물 스크리닝 모두에 유용함을 보여주었습니다.
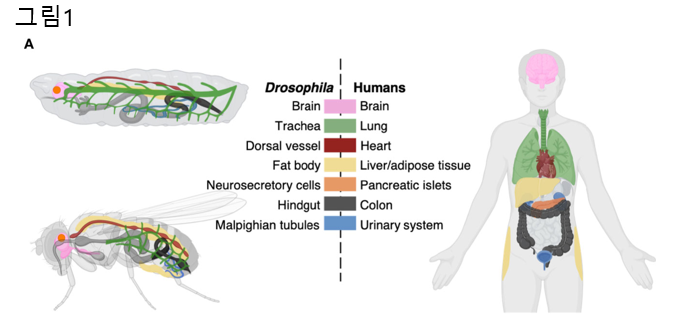
그림1. 초파리와 인간 장기의 구조 및 기능적 유사성. 초파리는 뇌, 지방체, 장기 등 주요 기관이 인간과 기능적으로 유사하여 암과 대사질환 연구에 적합한 모델 생물체임. 이러한 유사성은 인간 질환을 모사하는 다양한 파리 모델 구축을 가능하게 합니다.
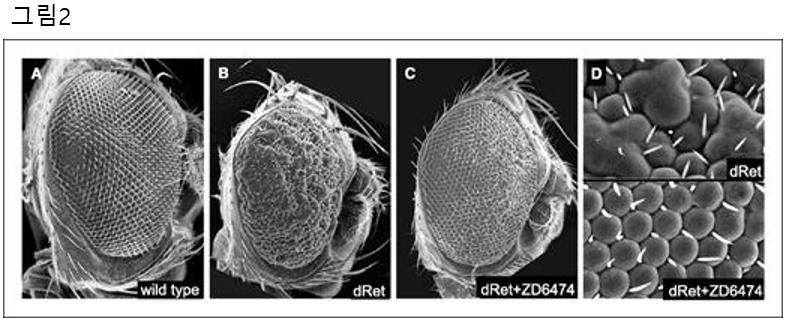
그림 2. 초파리 형질전환체를 이용한 RET 변이 암 모델
A. 야생형 초파리(wild type)의 정상적인 눈 구조.
B. Gal4/UAS 시스템을 통해 인간의 종양 유발 RET 변이(dRet)를 발현시킨 초파리에서 관찰되는 과도한 세포 증식과 형태 이상.
C. dRet 발현 초파리에 RET 저해제 ZD6474를 처리했을 때 비정상적 안구 발달이 억제되며, 정상에 가까운 구조가 회복되었습니다.
D. 고배율 주사전자현미경 이미지. dRet 발현에 의해 세포 배열과 형태가 붕괴되는 반면, ZD6474 처리 시 세포 간 배열이 회복됨을 보여줍니다.